
Last year I watched a 40-person biotech spend $180K on an enterprise QMS they used for exactly one thing: storing SOPs in folders. They could have done that with Google Drive and a spreadsheet.
The FDA compliance software market is confusing on purpose. Vendors lump together document management, submission tracking, quality systems, and AI compliance checking under one vague label ("regulatory compliance platform") and charge you $50K before you figure out which category you actually need.
I spent three weeks digging through G2 reviews, product demos, pricing pages (when they exist), and Crunchbase filings for 12 tools that biotech companies actually consider when preparing for FDA submissions. Some cost $180K/yr. One is free. Most fall somewhere in between.
Disclosure up front: We built Regfo, so we're biased. We've tried to be fair in this review. Our cons are listed too. We've placed ourselves in the category where we actually belong rather than claiming we're #1 at everything. If you spot something unfair, email me.
Before You Choose: QMS vs RIM vs Gap Analysis
People search "FDA compliance software" and get results mixing three very different product categories. Before you compare pricing, make sure you're comparing the right type of tool.
Quality Management Systems (QMS) handle document control, CAPAs, deviations, training records, and audit management. They're the backbone of your quality infrastructure. If FDA auditors walk in, they want to see your QMS. Think: Qualio, MasterControl, Greenlight Guru.
Regulatory Information Management (RIM) platforms track submissions, manage regulatory intelligence across markets, and organize dossiers. If you're filing in 30 countries simultaneously, you need RIM. Think: Veeva Vault, Kivo, RegDesk.
AI Gap Analysis tools read your actual study documents and protocols, compare them against FDA/ICH requirements, and tell you what's missing before you submit. They don't store your SOPs or track your CAPAs. They answer one question: "Is this submission-ready?" Think: Regfo, Weave Bio.
Here's the decision shortcut:
- Need to manage SOPs, training, and CAPAs for FDA audits? You need a QMS.
- Filing submissions in multiple countries? You need RIM.
- Want to know if your preclinical studies actually meet ICH requirements before you submit? You need gap analysis.
- Series B+ pharma with 200+ people? You probably need all three.
Most Series A-C biotechs need a QMS first, then gap analysis. RIM comes later when you're filing globally.
How We Evaluated
I looked at publicly available information for each tool:
- G2 and Capterra reviews. Real user feedback, not vendor marketing. Where reviews exist, I've quoted them directly.
- Pricing transparency. Does the vendor publish prices, or is it "contact sales" with a 3-week demo cycle?
- Setup time. Can you start this week, or does implementation take 6 months?
- Company fit. A tool built for 5,000-person pharma won't work for a 15-person biotech, and vice versa.
- ICH/FDA coverage. Does the tool actually understand regulatory requirements, or is it just document storage with a compliance label?
I did not test every tool hands-on. For tools without public reviews (Weave Bio, DIP-AI, Complizen), I relied on their marketing materials, press releases, and funding announcements. Take those sections with extra skepticism.
Enterprise QMS Platforms
If you're a top-20 pharma company reading this for some reason, these two are your options. Everyone else can skip to the next section, but read anyway so you understand what your $180K/yr competition is working with.
1. Veeva Vault QualityOne
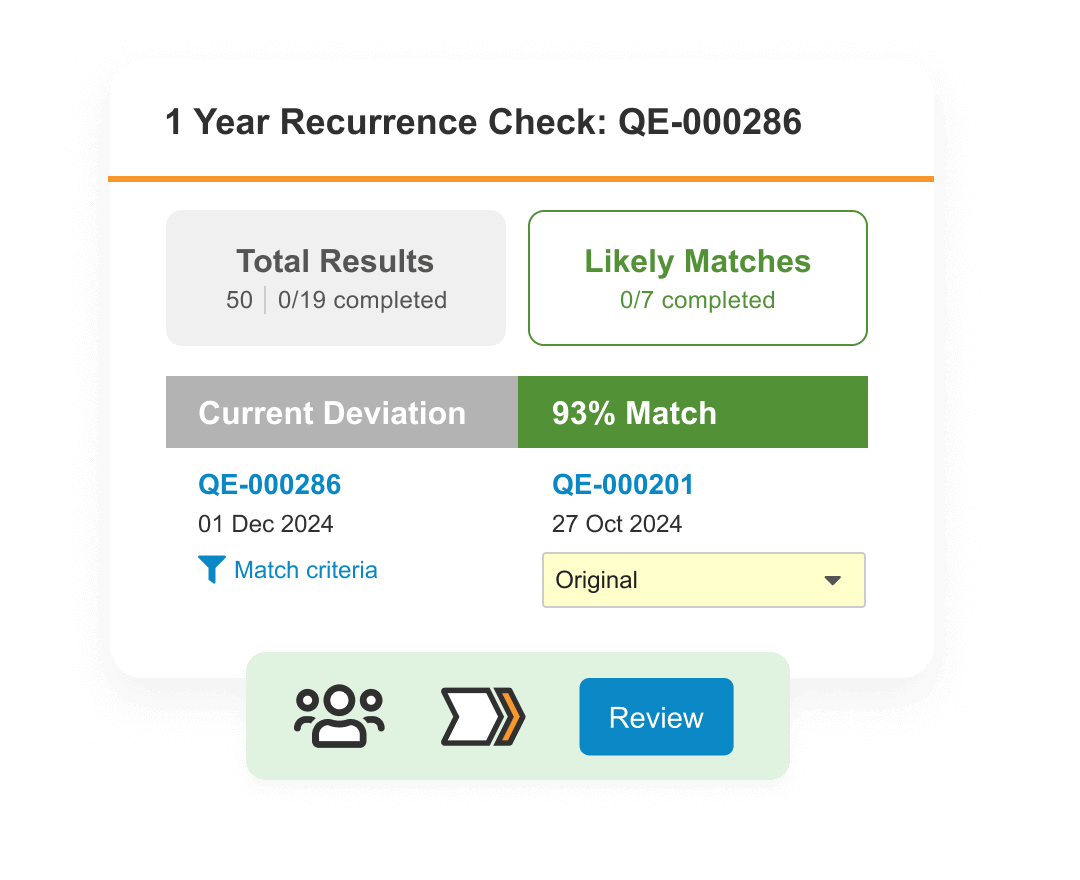
What it is: The 800-pound gorilla of life sciences software. Veeva is a public company (NYSE: VEEV) with $3.2B in annual revenue, 7,291 employees, and a cloud platform that covers everything from quality management to clinical data to regulatory submissions. Vault QualityOne is their quality and document management module.
Best for: Top-20 pharma companies with dedicated quality departments and six-figure software budgets.
Pricing: No public pricing. Enterprise deployments run $60K-180K/yr for 10 users, with implementation costs of $10K-50K for smaller deployments and significantly more for global rollouts. Individual user licenses range $50-200/month.
Pros:
- Industry standard. When FDA auditors see Veeva, they nod. It's table stakes for large pharma.
- Full lifecycle coverage. QMS, RIM, clinical, and regulatory in one platform.
- Ease of use rated 9.4/10 on G2, once you learn it.
- Responsive support team gets consistently positive reviews.
Cons:
- The price tag eliminates every startup, most mid-size biotechs, and probably your entire seed round.
- Steep onboarding curve. G2 reviewers note: "First users need to be trained, though continuous use provides better experience." Translation: it takes weeks before your team stops calling IT.
- PDF rendering issues. Documents have rendering problems when generating PDFs, per multiple reviews.
- Restrictive permissions. Only document owners can start workflows; owners can only change when status is "draft" or "effective." For small teams wearing multiple hats, this rigidity is painful.
- Implementation timelines of 6-12 months mean you won't see value until next year.
Verdict: If you have the budget and the team size to justify it, Veeva is the safe choice that no one gets fired for buying. If you're a 20-person biotech wondering whether to spend your Series A on Veeva, the answer is no.
2. MasterControl
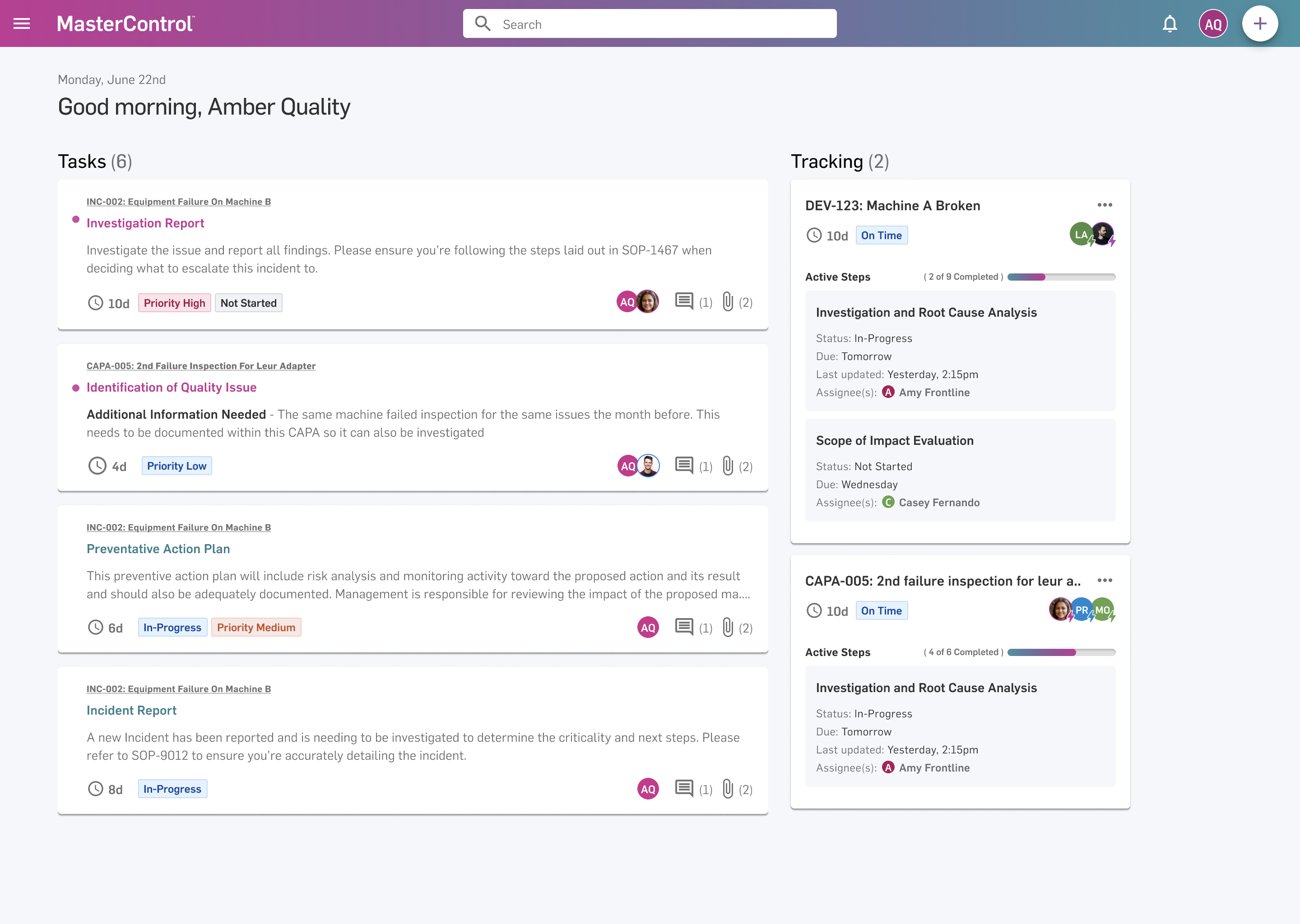
What it is: A full-stack QMS platform with $200M+ ARR and 667 G2 reviews. Document control, training management, CAPA, deviations, the full quality system. Backed by $150M in funding at a $1.3B valuation.
Best for: Mid-to-large pharma and biotech companies (200+ employees) with dedicated quality teams who need validated document control.
Pricing: Starts around $25K-30K/yr for 10 users, plus implementation costs. Named-user licensing. No public pricing page.
Pros:
- Covers every QMS module you'd need: document control, training, CAPA, deviations, change control all in one system.
- Strong validation documentation (IQ/OQ/PQ) for FDA audit readiness.
- 667 G2 reviews (4.4/5), one of the most reviewed tools in this list.
- Good training management module that integrates with document workflows.
Cons:
- The interface is the #1 complaint. From G2: "The interface is non-intuitive as users struggle with navigation and terminology."
- Bugs are a recurring issue. One reviewer wrote: "They have 'bugs' that are often not fully vetted until a customer points them out." That's not a great sign for a quality system.
- Finding people who can administer it is hard: "Finding competent admins is a potential issue — the setup is not straightforward and not everyone can wrap their head around the system."
- Reporting is clunky. Limited functionality, unintuitive interfaces, and unreliable standard reports.
- eForms and document processing features require significant manual administration despite being positioned as automated.
Verdict: MasterControl does what it says. It's a serious, validated QMS. But the interface complaints are loud and consistent across 667 reviews. If your quality team has the patience for a learning curve, it'll serve you well. If you need something your scientists can actually use without 3 days of training, keep reading.
Small Biotech QMS & RIM
These tools target the companies that can't afford Veeva but still need real quality infrastructure. The price range here is $12K-35K/yr. Still not cheap, but within reach for funded startups.
3. Qualio
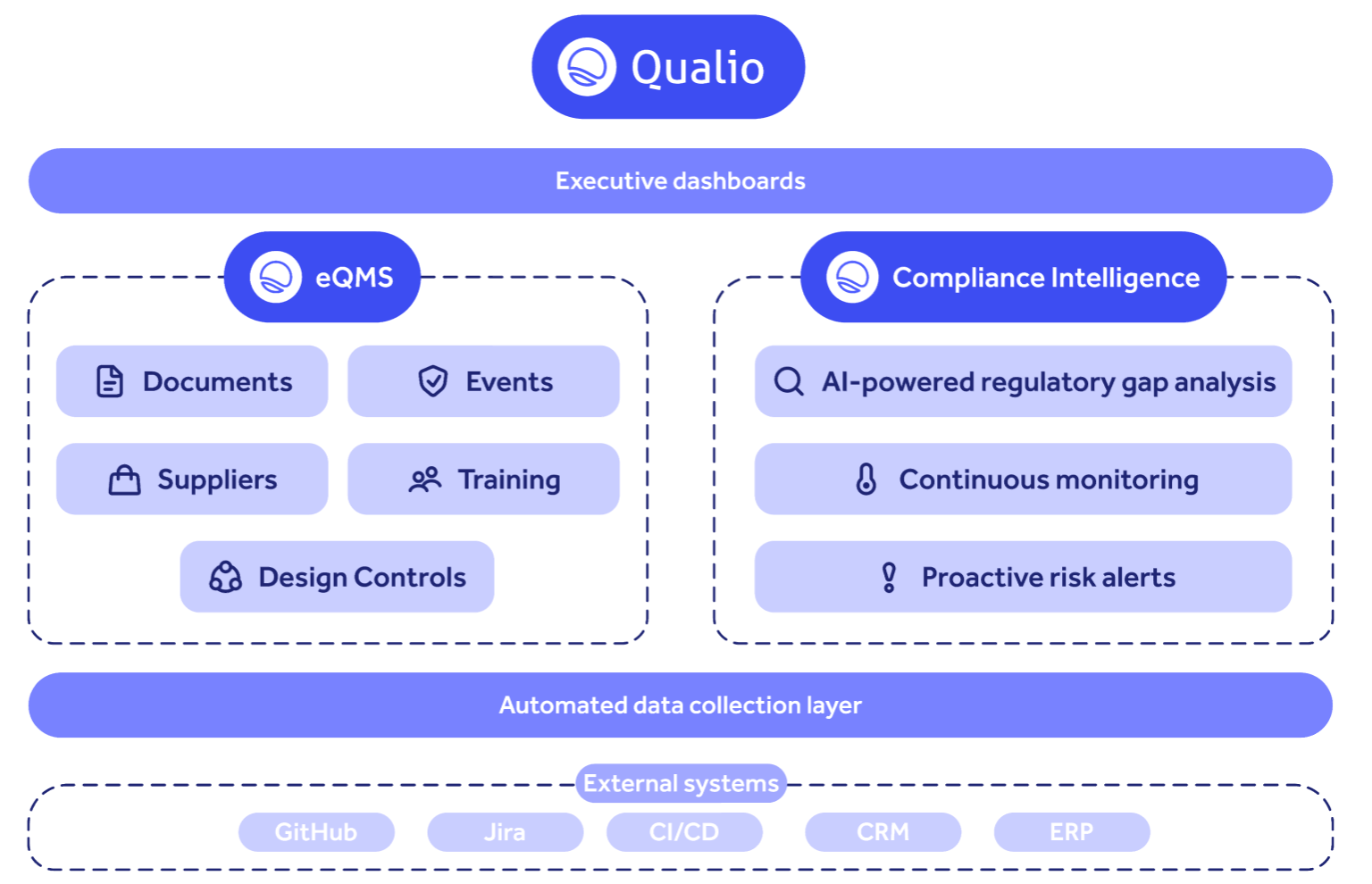
What it is: Cloud-based eQMS built for small to mid-size life sciences companies. 682 G2 reviews, $63.7M in funding (Series B from Menlo Ventures and Tiger Global), 500+ customers across 80 countries. Qualio's pitch is "easiest eQMS for life sciences," and the review data mostly backs that up.
Best for: Series A-C biotechs (10-200 employees) that need a QMS up and running in weeks, not months.
Pricing: Starts around $12K/yr. Six subscription tiers. Generally affordable for small companies, but can get expensive at scale. Quote-based, so you'll need to talk to sales.
Pros:
- Ease of use rated 8.9/10 on G2. Your team will actually use this without begging.
- Document control rated 9.2/10, their strongest feature.
- Support rated 9.2/10. When something breaks, people answer.
- Claims 5X ROI within 2 months and 99% reduction in quality admin time. Take vendor ROI claims with salt, but the review scores are real.
- Setup takes weeks, not months.
Cons:
- The document editor is a known weak spot. From G2: "The document editor is not as robust as Microsoft Word. This makes custom documents harder to create."
- Scales poorly. Another reviewer: "We have so many saved documents it sometimes becomes cumbersome to remember where to find things."
- Limited audit trail. You can't view detailed comment history, and change control shows only the last change. That's a problem during FDA inspections.
- The document creation workflow is still rough: "Qualio still have some sticking points with the document creation journey... they are still sorely lacking ease with the creation, reviewer and approval experience."
Verdict: Qualio is probably the best QMS choice for a 20-50 person biotech. It's affordable, it's usable, and it's got the review volume to prove it works. Just don't expect the document editor to replace Word. You'll still be drafting outside the platform and importing.
4. SimplerQMS
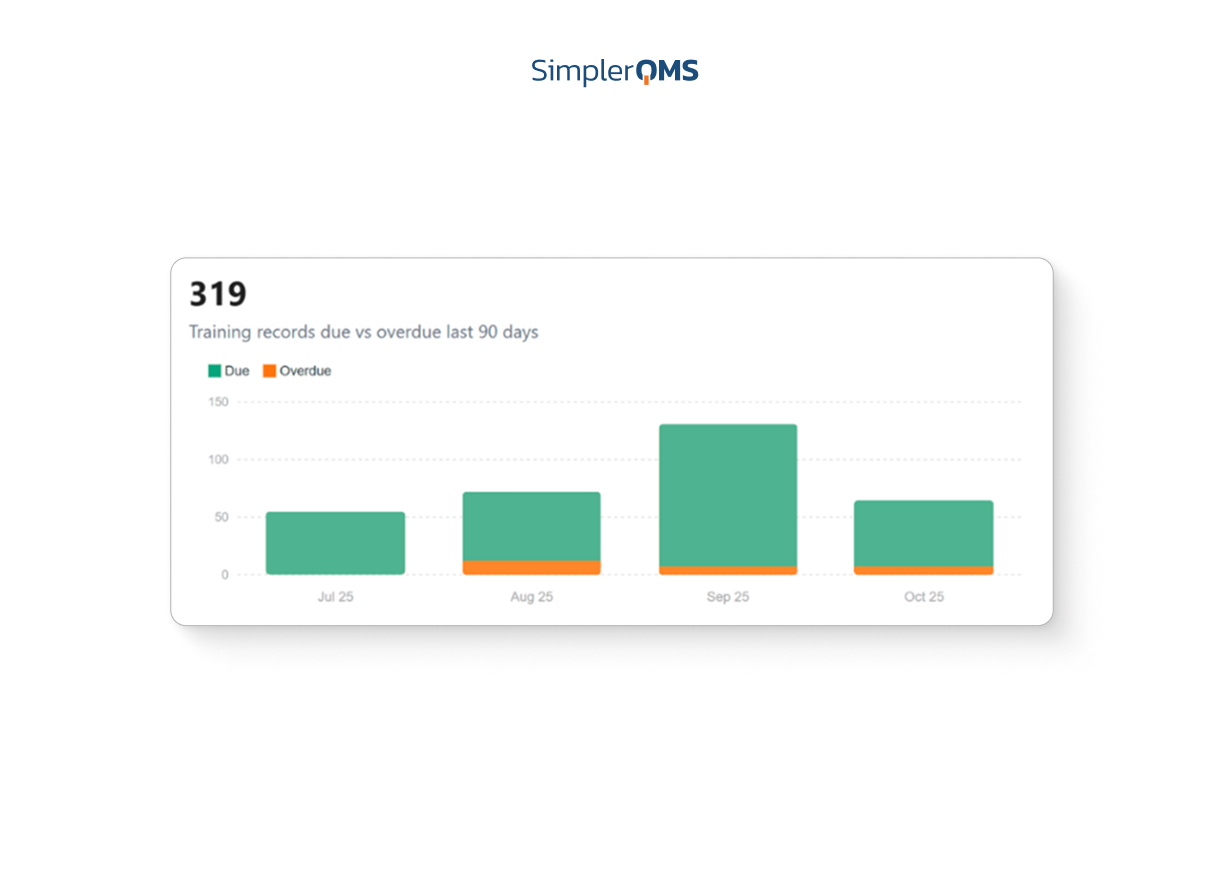
What it is: A smaller, bootstrapped eQMS (EUR 4M raised, 34 employees) built on top of M-Files. Pre-configured templates, included validation, and competitive pricing for companies that want quality infrastructure without the enterprise overhead.
Best for: Budget-conscious small biotechs that want a pre-configured QMS with validation included in the price.
Pricing: Starts at $13,750/yr. Per-user yearly subscription. Includes implementation, validation, updates, unlimited training, hosting, templates, and 24/7 support. Free Viewer licenses available.
Pros:
- All-inclusive pricing. Validation, templates, training, and support included. No hidden implementation fees.
- Pre-configured for life sciences. You're not starting from a blank platform.
- Responsive support team gets consistently positive feedback.
- Competitive pricing compared to MasterControl or Qualio for very small teams.
Cons:
- Built on M-Files, and the underlying platform shows. From G2: "M-Files interface can be unstable sometimes, resulting in freezing or certain functions like approving documents to become buggy, requiring a reboot."
- The e-signatures are a deal-breaker for some: "Electronic signatures are displayed very inefficiently and add 3 pages to documents." Three pages of signature blocks on every document. In 2026.
- Limited reporting. Users struggle to generate reports and harness metadata.
- One independent reviewer characterized the UI as having "a UI implementation of Windows 95." Harsh, but the screenshots don't disagree.
- Onboarding is brief and doesn't cover all functionality. Users report being left to figure out modules on their own.
Verdict: SimplerQMS offers real value for very small teams. The all-inclusive pricing is genuinely appealing when competitors charge separately for implementation, validation, and training. But the M-Files foundation creates friction, and the dated interface is hard to ignore. If UI matters to your team (it should), this one will feel like a step backward.
5. Greenlight Guru

What it is: The only QMS built specifically for medical devices. 401 G2 reviews, $121M in funding, and a purpose-built platform with CAPA requirements, design controls, and risk management for med dev. If you're doing a 510(k) or PMA, this is designed for you.
Best for: Medical device companies. Not pharma, not biotech doing IND submissions. Devices.
Pricing: Starts around $15K/yr; typical deployments cost $25K-35K/yr for small companies. Warning: prices doubled in January 2026, and multiple G2 reviewers are furious about it. Minimum 2-3 year contracts. ITQlick rates their pricing value at 2.4/10.
Pros:
- Purpose-built for medical devices. Design controls, risk management, CAPA all structured around ISO 13485 and FDA 21 CFR 820.
- Intuitive interface that centralizes essential QMS functions for device teams.
- Fast implementation: 3-5 months including training.
- 401 G2 reviews at 4.5/5. Strong track record with device companies.
Cons:
- Pricing is the #1 complaint, and it's recent. From G2 after the January 2026 price increase: "Their platform underdelivered at every turn: clunky, counterintuitive, and nowhere near as efficient as advertised."
- Customer service has deteriorated. Multiple reviewers report: "The customer service experience feels impersonal — every representative we spoke with sounded like they were reading from the same script, using identical phrasing."
- One reviewer's summary: "Too expensive, terrible customer service and attention."
- The strongest complaint: "Their rigid stance, complete lack of empathy, and unhelpful customer service made the entire ordeal a nightmare."
- Search functionality fails to yield relevant results. Analytics provides limited useful data.
- Not designed for pharma IND submissions. The entire structure is oriented around device regulations, not ICH guidelines.
Verdict: A year ago I would have called Greenlight Guru the clear winner for device companies. The January 2026 price doubling changed the equation. If you're already locked into a contract, you're fine. If you're evaluating new QMS tools for a device company, compare the new pricing carefully against Qualio and Kivo before signing a 3-year deal. If you're a biotech doing IND submissions, this is the wrong tool entirely.
6. Kivo

What it is: An all-in-one platform combining RIM, eTMF, and QMS for emerging life sciences companies. 17 G2 reviews at a perfect 5.0/5 rating (small sample size, but the consistency is notable). Kivo includes everything without separate module charges.
Best for: Small to mid-size biotech companies preparing their first IND submission who want RIM + eTMF + QMS in one platform.
Pricing: Starts at $1,800/mo (~$21,600/yr). Enterprise pricing around $85K/yr for 100 users. One-time implementation starting at $25K. All features included. No per-study or per-GB charges.
Pros:
- All-in-one: RIM, eTMF, and QMS included without separate charges. Competitors sell these as three different products.
- Fully validated, Part 11 and SOC 2 compliant out of the box.
- Strong document organization with metadata tagging and version control.
- Built for emerging biotech, not retrofitted from enterprise tools.
- 5.0/5 on G2, the only tool in this list with a perfect score (though only 17 reviews).
Cons:
- Navigation can be confusing. From G2: "Difficult to start working on the file. When we had a short timeline, starting to write is extremely important. It took me about 1 hr to figure out how to start."
- Limited project management features. Users want more PM and tracking capabilities.
- Relatively small review base (17 reviews). Harder to assess long-term reliability.
- $25K implementation fee on top of subscription costs adds up for early-stage companies.
Verdict: Kivo is underrated. The all-in-one approach means you're not buying three separate tools and trying to make them talk to each other. The 5.0 G2 rating is impressive even with the small sample. The main risk is company size. With funding not publicly disclosed, the longevity question is worth asking during the sales process. For a biotech preparing its first IND that needs regulatory infrastructure (not just gap analysis), Kivo is worth a serious look.
AI-Powered Regulatory Tools
These are the newer entrants using AI to automate regulatory work rather than just managing documents. They're earlier stage, have fewer reviews, and carry more risk. But they solve problems that traditional QMS tools don't even attempt.
7. Weave Bio

What it is: AI-native regulatory platform focused on automating the IND submission process. $36M in total funding ($20M Series A), Parexel partnership, and a small team of around 30 people. Their pitch: accelerate regulatory timelines by 50%+ using AI to handle eCTD formatting, data organization, authoring, review, and publishing.
Best for: Well-funded biotechs (Series B+) that want end-to-end IND automation and have the budget for enterprise AI tooling.
Pricing: Not public. Enterprise/custom pricing. If you have to ask, it's probably more than $50K/yr.
Pros:
- AI-native from day one, not a legacy QMS with AI bolted on.
- AutoIND module covers the full IND assembly pipeline: data organization, authoring, review, publishing.
- Parexel partnership adds validation credibility in a space where trust matters.
- Covers FDA, EMA, and global regulatory standards.
- Strong funding ($36M) from USVP and Innovation Endeavors.
Cons:
- No public user reviews. Zero G2 reviews. Zero Capterra reviews. You're trusting marketing materials and press releases.
- Enterprise pricing likely puts this out of reach for seed and Series A companies.
- ~30 employees means limited support bandwidth during your critical submission timelines.
- Broad scope (full lifecycle) may mean less depth in any single area, though this is speculation without user data.
- No free tier or self-serve option. You can't try before committing to a sales cycle.
Verdict: Weave Bio is solving the right problem. The Parexel partnership is the strongest credibility signal here. But with zero public reviews, you're an early adopter, and early adoption in regulated industries carries real risk. If you're well-funded and comfortable being a design partner, it's worth a conversation. If you need proven reliability for your first IND, the lack of track record is hard to ignore.
8. Ketryx
.png)
What it is: AI-powered quality and compliance platform focused on software-as-a-medical-device (SaMD) and connected medical devices. $57M in total funding ($39M Series B), trusted by 4 of the top 5 medical device manufacturers. Ketryx lets development teams stay in their existing tools (Jira, GitHub, etc.) while generating compliance documentation automatically.
Best for: Medical device and SaMD companies that need IEC 62304, ISO 13485, and FDA 21 CFR Part 11 compliance without leaving their dev tools.
Pricing: Per-user subscription, tiered by company stage (pre-market, early-stage, scaling). No minimum commitment, monthly pricing, flexible contracts. Specific prices not public.
Pros:
- Reduces documentation burden by up to 90%, a claim backed by their enterprise customer base.
- Teams keep working in Jira, GitHub, and existing dev tools. No workflow disruption.
- Trusted by 4 of top 5 medical device manufacturers. Strong enterprise validation.
- Broad compliance coverage: IEC 62304, FDA 21 CFR Part 11, ISO 13485, EU MDR.
- Flexible contracts. No multi-year lock-ins.
Cons:
- Only 1 G2 review. Not enough data to assess real-world user experience.
- Firmly focused on device/SaMD compliance. If you're developing a small molecule or biologic for an IND submission, this isn't your tool.
- Per-user pricing without published rates makes budgeting difficult.
- At 62-97 employees with $6.8M revenue, the ratio suggests they're still burning cash. Worth considering for long-term vendor stability.
Verdict: If you're building software-as-a-medical-device and your engineering team lives in Jira and GitHub, Ketryx is the most natural fit in this list. The enterprise customer base (4 of top 5 device manufacturers) is strong validation. For pharma and biotech companies doing IND submissions: wrong product, right idea. Keep your eye on them in case they expand scope.
9. DIP-AI (Deep Intelligent Pharma)
What it is: Singapore-based AI platform covering the full clinical trial workflow: protocols, clinical study reports, investigator's brochures, IND/IMPD submissions. 1,000+ pharma/biotech clients including Bayer, J&J, and Roche. $165M+ in total funding.
Best for: Large pharma companies with Asia-Pacific operations that need AI-powered eCTD publishing and multi-language clinical document automation.
Pricing: Enterprise/custom. Not publicly available.
Pros:
- Massive client base: 1,000+ pharma/biotech clients, including top-10 pharma names.
- Full clinical trial document automation: protocols, CSRs, IBs, submissions.
- Multi-language support with AI translation, which matters for global filings.
- $165M+ in funding signals serious backing and longevity.
- Claims "up to 1000% efficiency gains and 99% accuracy" on document processing.
Cons:
- No G2 reviews. No Capterra reviews. No Western review platform presence at all.
- Primarily serves Asia-Pacific market. Western market penetration and support are unclear.
- "1000% efficiency gains" is the kind of claim that makes regulatory professionals squint.
- Enterprise pricing with no transparency.
- China-headquartered (Singapore HQ, Beijing offices). Some companies have data residency concerns for regulatory documents.
Verdict: DIP-AI has impressive funding and client logos, but the lack of any Western review data makes independent assessment impossible. If you're a pharma company with APAC operations, it's worth exploring. For US/EU-focused biotechs, the other options on this list have better verifiable track records in Western regulatory environments.
The Preclinical Gap Analysis Approach
Here's the gap none of the tools above fill.
QMS tools store your documents. RIM tools track your submissions. But neither reads your actual preclinical study reports and tells you whether they meet ICH requirements before you submit to FDA.
Think about what happens during IND preparation. You've run 8-12 preclinical studies. Each one needs to comply with specific ICH guidelines: safety pharmacology under ICH S7A, genotoxicity under ICH S2, repeat-dose tox under ICH M3(R2). Your regulatory consultant manually cross-references each study against these requirements, flags gaps, and charges $300-500/hour for the privilege.
That review takes weeks. Sometimes months. And if they miss something, you find out when FDA issues a clinical hold.
Gap analysis tools automate that cross-referencing. They read the content of your studies, compare against regulatory requirements, and tell you what's missing.
10. Regfo
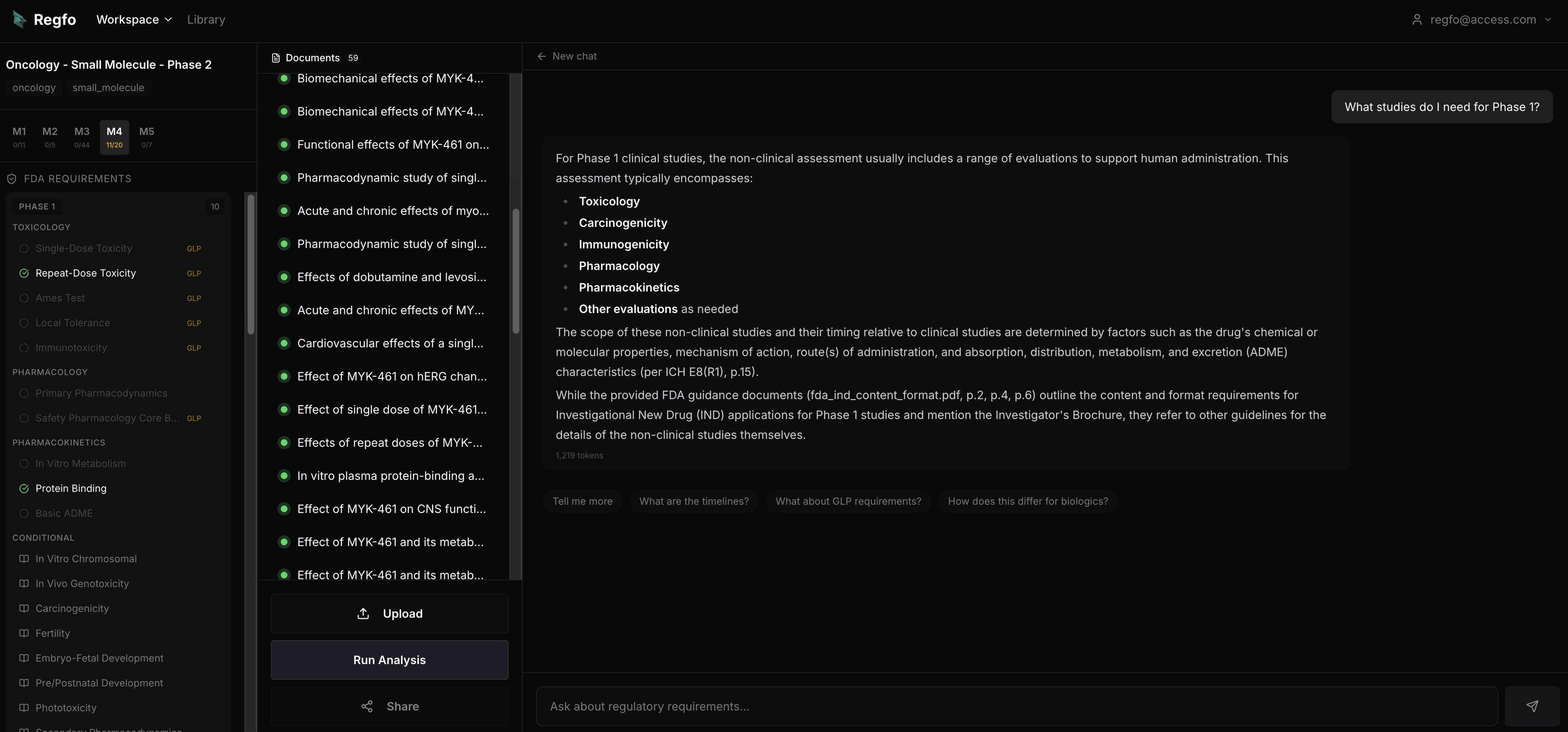
What it is: AI-powered regulatory gap analysis tool that checks preclinical study packages against ICH/FDA requirements. Upload your studies, configure your study package, and get a compliance score with specific guideline citations. Built for biotech teams preparing IND submissions, not for managing SOPs or tracking CAPAs.
Best for: Series A-C biotechs (10-50 employees) preparing IND submissions who want to find compliance gaps before consultants or FDA do.
Pricing: Free tier (limited). Pro: $399/mo. Team: $999/mo. Prices are on the website. No "contact sales" dance.
Pros:
- Checks actual study content against 24 ICH guidelines, covering nonclinical study types from safety pharmacology to carcinogenicity.
- Every finding cites a specific guideline section. ICH S7B Section 2.1, not "consider cardiac safety."
- Compliance score in under 30 seconds. Not days, not weeks.
- Free tier lets you test with real data before paying.
- Transparent pricing. $399/mo is roughly what a regulatory consultant charges for one hour.
- Free CTD Library covering 218 sections for reference.
Cons:
- Focused on preclinical and IND. If you need post-market surveillance, GMP compliance, or device regulations, this isn't it.
- Not a QMS. No document control, no CAPA management, no training records. You'll still need Qualio or similar for that.
- Newer product with less track record than MasterControl (667 reviews) or Qualio (682 reviews). We're building that trust, but we're not there yet.
- Currently strongest coverage for small molecule programs. Biologics and cell/gene therapy coverage is expanding but not as deep.
- AI gap checking means results need human review. This is a co-pilot, not autopilot. Your regulatory lead still makes the final call.
Verdict: I'm obviously biased here, but I'll be specific about what Regfo is and isn't. It's the fastest way to check whether your preclinical package meets ICH requirements before submission. It's not a replacement for your QMS, your regulatory consultant, or your RA team. Think of it as the spell-checker that catches the obvious gaps so your consultants can focus on strategy instead of cross-referencing PDFs.
If you're a 15-person biotech spending $40K on consultant reviews to find out your genotoxicity battery is missing an in vivo assay, you could have caught that in 30 seconds for $399/mo. That's the pitch. Try it free.
Niche & Specialized Tools
These two tools serve specific use cases that might be exactly what you need, if you're in their target market.
11. Complizen
What it is: Early-stage AI tool focused on FDA compliance for medical devices, specifically 510(k) submissions. Free tier for startups, with a paid Plus tier for additional features. Built by Zen Labs Inc.
Best for: Medical device startups navigating their first 510(k) submission.
Pricing: Free tier available. Plus tier is per-seat with monthly or annual billing, but specific pricing is not publicly disclosed. Annual discounts available.
Pros:
- Free tier lets you explore before paying. Rare in this space.
- AI-powered regulatory requirement retrieval for 510(k) pathways.
- Claims to save 12+ hours/week on regulatory research.
- 510(k) submission roadmap generation can help first-time submitters understand the process.
Cons:
- Zero G2 reviews. Zero Capterra reviews. Very early stage.
- Medical device only. No utility for pharma or biotech IND submissions.
- Team size, funding, and company stability unknown. Vendor risk is real.
- "Saves 12+ hours/week" is a marketing claim without published validation.
- Plus tier pricing not publicly disclosed for a product that offers a free tier. Seems like an oversight or a sign of early-stage pricing uncertainty.
Verdict: Interesting concept for device startups, but too early to recommend with confidence. The free tier means you can try it without risk. If you're doing a 510(k) and want AI assistance, give it a spin. But don't rely on it as your only compliance tool until there's real user validation.
12. RegDesk
-1.png)
What it is: AI-powered Regulatory Information Management platform focused on medical devices across 120+ countries. ~7 G2 reviews at ~4.5/5. RegDesk helps MedTech companies understand regulatory requirements across jurisdictions. If you need to know what Japan's PMDA requires vs. the EU MDR vs. FDA, this is designed for that.
Best for: Medical device companies managing submissions across multiple international markets.
Pricing: Not publicly available. Contact sales. Likely premium-priced given the multi-market regulatory intelligence they provide.
Pros:
- Regulatory intelligence covering 120+ countries. The broadest geographic coverage in this list.
- AI tools for document translation and regulatory requirements.
- Team is professional and responsive, fostering strong partnerships per G2 reviews.
- Platform evolves with industry changes, which matters in a regulatory landscape that shifts constantly.
Cons:
- Reporting is limited. From G2: "Reporting is not always easy to access as an end-user, and the data upload process was lengthy and somewhat manual."
- Manual data upload process. Challenges with bulk data can confuse internal teams.
- No public pricing. The "contact sales" wall adds friction for small teams.
- MedTech focused. Limited utility for pharma and biotech IND workflows.
- Only 7 G2 reviews. Small sample for a company founded in 2014.
Verdict: If you're a device company filing in 20+ countries, RegDesk's multi-market intelligence is genuinely valuable and hard to replicate manually. For a US-only biotech preparing a single IND, the geographic coverage is irrelevant and the price likely isn't justified. Know your use case before scheduling the demo.
Quick Comparison Table
| Product | Category | Best For | Pricing (Annual) | Free Tier | Setup Time | G2 Rating | Reviews |
|---|---|---|---|---|---|---|---|
| Veeva Vault | Enterprise QMS+RIM | Top-20 pharma | $60K-180K+ | No | 6-12 months | 4.1-4.5/5 | 24 |
| MasterControl | Enterprise QMS | Large pharma/biotech | $25K-30K+ | No | 3-6 months | 4.4/5 | 667 |
| Qualio | Cloud QMS | Small-mid biotech | ~$12K+ | No | Weeks | 4.4/5 | 682 |
| SimplerQMS | Cloud QMS | Budget biotech | $13,750+ | No | Weeks | ~4.5/5 | 5 |
| Greenlight Guru | Device QMS | Medical devices | $25K-35K+ | No | 3-5 months | 4.5/5 | 401 |
| Kivo | RIM + QMS | Emerging biotech | $21,600+ | No | Weeks | 5.0/5 | 17 |
| Weave Bio | AI regulatory | Well-funded biotech | Enterprise | No | Weeks | N/A | 0 |
| Ketryx | AI quality | SaMD/device | Per-user | No | Weeks | N/A | 1 |
| DIP-AI | AI docs | Large pharma (APAC) | Enterprise | No | Weeks | N/A | 0 |
| Regfo | AI gap analysis | Preclinical teams | $4,788/yr | Yes | Minutes | N/A | New |
| Complizen | AI compliance | 510(k) devices | Per-seat | Yes | Minutes | N/A | 0 |
| RegDesk | Reg intelligence | Multi-market devices | Enterprise | No | Weeks | ~4.5/5 | 7 |
A few things jump out from this table.
The tools with the most reviews (MasterControl at 667, Qualio at 682) are the cloud QMS platforms. That's where the market maturity is. The AI-powered tools are almost all pre-review. You're comparing proven-but-older against promising-but-unproven.
Only two products offer a free tier (Regfo and Complizen). Only one publishes its pricing on the website without a "contact sales" button (Regfo). That's unusual for this market and says something about who each vendor considers their customer.
If you're a Series A biotech preparing for IND and you want to see where Regfo fits, upload a study and get a free compliance score →
How to Choose the Right Tool
Skip the feature comparison spreadsheets. Answer these questions instead.
What's your company size?
- 500+ employees, $50M+ revenue → Veeva or MasterControl. You need enterprise infrastructure, and you can afford it.
- 50-500 employees → Qualio, Kivo, or MasterControl depending on budget and whether you need QMS alone or QMS + RIM.
- 10-50 employees → Qualio or SimplerQMS for QMS. Regfo for gap analysis. Kivo if you need all-in-one.
- Under 10 employees → Free tools first. Regfo's free tier for gap analysis. Complizen's free tier for device 510(k).
What are you building?
- Small molecule or biologic for IND → Qualio (QMS) + Regfo (gap analysis). Add Kivo if you need RIM.
- Medical device → Greenlight Guru or Ketryx. Add RegDesk if filing internationally.
- Software-as-medical-device → Ketryx. It integrates with your dev tools.
- Cell/gene therapy → Qualio (QMS) + consultant. The AI tools haven't caught up to CGT-specific requirements yet.
What's your immediate problem?
- "We need to manage SOPs, CAPAs, and training" → QMS first. Qualio or SimplerQMS.
- "We need to know if our preclinical package is IND-ready" → Gap analysis. Regfo.
- "We're filing in 15 countries" → RIM. RegDesk or Kivo.
- "We're assembling the actual IND dossier" → Weave Bio or DIP-AI, if you have the budget for enterprise AI.
- "We need everything" → Kivo (combined) or Qualio + Regfo (modular).
What's your budget?
- Under $5K/yr → Regfo Pro ($4,788/yr), Complizen free tier, or manual processes with the free CTD Library.
- $5K-25K/yr → Qualio, SimplerQMS, or Kivo.
- $25K-100K/yr → MasterControl, Greenlight Guru, or Kivo enterprise.
- $100K+/yr → Veeva. Full stop.
FAQ
What's the difference between QMS and regulatory compliance software?
A QMS (Quality Management System) manages your quality infrastructure: document control, SOPs, CAPAs, training records, deviations, and audit management. It's what FDA inspectors want to see when they walk in. Tools like Qualio, MasterControl, and Greenlight Guru are QMS platforms.
"Regulatory compliance software" is a broader, vaguer term that vendors use to mean different things. It might mean a QMS, or it might mean regulatory intelligence (tracking requirements across markets), or it might mean gap analysis tools that check whether your actual study data meets FDA requirements. Before you compare products, figure out which of these three things you need. Most Series A-C biotechs need a QMS first, then gap analysis for their specific submission.
Do I need FDA compliance software for IND submission?
You don't technically need any software. Plenty of companies have submitted INDs using Word documents, Excel checklists, and a good regulatory consultant. The question is whether that's the best use of your time and money.
A typical IND preparation involves cross-referencing 8-15 preclinical studies against multiple ICH guidelines: M3(R2) for study timing, S7A/S7B for safety pharmacology, S2 for genotoxicity, S6 for biologics. Doing this manually takes weeks and costs $10K-50K in consultant time. A gap analysis tool does it in minutes for a fraction of the cost. You decide if the math works for your burn rate.
Can I use free tools for FDA compliance?
Yes, with caveats. Regfo offers a free tier that lets you explore compliance checking with limited usage. Complizen has a free tier for 510(k) device submissions. Beyond purpose-built tools, the FDA and ICH publish all their guidance documents for free. The challenge is finding, reading, and cross-referencing 100+ page PDFs against your specific studies by hand.
Free tools won't replace a QMS if you need document control for FDA audits. And free tiers typically limit the number of analyses or documents you can process. They're a starting point, not an endpoint. For a Series A biotech with limited budget, starting free and upgrading once you've validated the tool's value is the smart approach.
How much does FDA compliance software cost?
The range is enormous. Here's the real breakdown from this review:
- Free: Regfo free tier, Complizen free tier
- $5K-15K/yr: Qualio (starts ~$12K), SimplerQMS ($13,750)
- $15K-35K/yr: Greenlight Guru ($25-35K post-price increase), Kivo ($21.6K)
- $25K-100K/yr: MasterControl ($25-30K+), Kivo Enterprise ($85K for 100 users)
- $60K-180K+/yr: Veeva Vault
- Enterprise (price unknown): Weave Bio, DIP-AI, Ketryx, RegDesk
Don't forget implementation costs. Veeva and MasterControl typically charge $10K-50K+ for setup. Kivo charges $25K. SimplerQMS includes implementation in the subscription. Cloud-native tools like Qualio and Regfo have minimal implementation costs.
What ICH guidelines does FDA check during IND review?
FDA reviewers assess your IND against a specific set of ICH guidelines depending on your drug type, modality, and clinical phase. The core ones for a first-in-human small molecule IND include:
- ICH M3(R2): Timing of nonclinical studies relative to clinical development
- ICH S7A/S7B: Safety pharmacology core battery and supplemental studies
- ICH S2(R1): Genotoxicity testing and data interpretation
- ICH S6(R1): Preclinical safety evaluation of biotechnology-derived products (for biologics)
- 21 CFR Part 58: GLP compliance requirements
FDA doesn't check every ICH guideline for every submission. The specific requirements depend on your drug modality, therapeutic area, and development phase. Our IND submission checklist covers the full mapping.
Is AI-generated compliance checking accepted by FDA?
FDA doesn't "accept" or "reject" compliance tools. They evaluate your actual submission data. Whether a human or AI found your gaps doesn't matter to FDA. What matters is that your studies meet the requirements.
AI compliance tools like Regfo, Weave Bio, and Ketryx are decision-support tools, not submission tools. They help your team find gaps before submission, the same way spell-check helps you find typos before sending an email. Your regulatory team reviews the AI findings, makes decisions, and takes responsibility for the submission. FDA never sees or evaluates the AI tool itself. The question isn't "will FDA accept AI" but "will AI help us catch issues that humans miss?" Based on how many common IND gaps are preventable with a pre-submission review, the answer is yes.
How long does it take to implement a QMS?
Implementation timelines vary dramatically by product:
- Veeva Vault: 6-12 months for full enterprise deployment
- MasterControl: 3-6 months with dedicated implementation team
- Greenlight Guru: 3-5 months including training
- Qualio: Weeks. Their fastest implementations are under 30 days
- SimplerQMS: Weeks. Pre-configured templates accelerate setup
- Kivo: Weeks for initial setup, though $25K implementation engagement
For AI tools like Regfo and Complizen, "implementation" is measured in minutes. Create an account, upload documents, get results. No configuration, no IT involvement, no implementation team.
The bigger question is how long until your team actually uses it daily. Even fast-to-implement tools take 2-4 weeks before they're embedded in workflows. Budget for training time regardless of what the vendor promises.
What's the minimum compliance infrastructure for a Series A biotech?
At Series A (typically $5-15M raised, 10-20 employees), you need enough infrastructure to not embarrass yourself at an FDA pre-IND meeting and to actually get your IND filed without a clinical hold. Here's the minimum:
- A QMS for document control. Qualio ($12K/yr) or SimplerQMS ($13,750/yr). You need version-controlled SOPs, training records, and basic CAPA management. FDA expects this.
- A gap analysis tool. Regfo free tier or Pro ($399/mo). Know what your preclinical package is missing before spending $30K on a consultant review.
- A regulatory consultant. Yes, still. Software doesn't replace the person who's filed 50 INDs and knows what the specific FDA division cares about. But software can make their time more efficient. Use the $300/hour on strategy, not on cross-referencing PDFs.
Total minimum: ~$17K/yr for QMS + gap analysis, plus consultant hours as needed. That's roughly 10% of what Veeva costs and covers 90% of what a Series A biotech actually needs.
Do medical device companies need different compliance software than pharma/biotech?
Yes, and the difference matters more than most comparison articles acknowledge. Device companies operate under FDA 21 CFR 820 (Quality System Regulation), ISO 13485, and EU MDR. Pharma and biotech operate under 21 CFR Parts 210/211 (cGMP), ICH guidelines, and different submission types (IND/NDA vs 510(k)/PMA).
Greenlight Guru and Ketryx are built specifically for devices. Qualio and MasterControl serve both but aren't specialized for either. Regfo is built for pharma/biotech IND submissions and won't help with your 510(k). Complizen targets device 510(k) submissions specifically.
Buying a pharma-focused QMS for a device company (or vice versa) means fighting your tool instead of using it. Pick the tool that matches your regulatory pathway.
If you're preparing an IND and want to check your preclinical package before a consultant does, try Regfo free → It takes 3 minutes to upload a study and see the gaps.
This article was last updated in April 2026. Pricing and feature information is based on publicly available data as of the publication date. If you spot an error, reach out and we'll correct it.